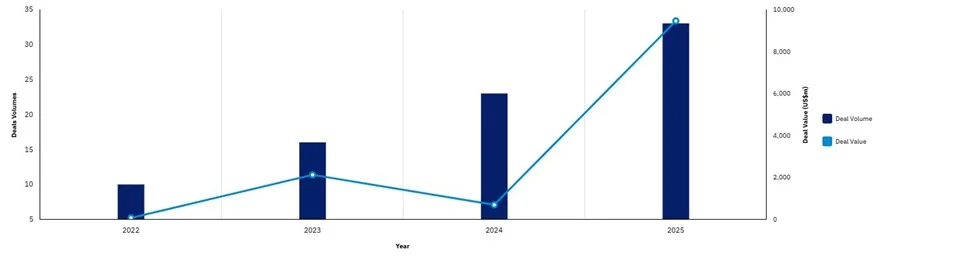
The FDA recently released draft guidance introducing a “plausible mechanismframework,” a new regulatory pathway designed to accelerate approval ofindividualized therapies for ultra-rare diseases. The framework allows sponsors torely on strong biological rationale, limited patient data, and alternative evidencesources (e.g., natural history or external controls) in place of traditionalrandomized clinical trials, particularly in settings where small patient populationsmake conventional trials infeasible.


The FDA recently granted approvals to two novel therapiestargeting ultra-rare pediatric diseases, highlighting continuedmomentum in gene and enzyme-based innovation. DenaliTherapeutics received accelerated approval for Avlayah, the firstnew treatment in ~20 years for Hunter syndrome and the firstenzyme therapy to cross the blood-brain barrier, addressingneurological symptoms based on biomarker endpoints.Meanwhile, Rocket Pharmaceuticals secured approval forKresladi, the first gene therapy for Leukocyte AdhesionDeficiency-I (LAD-I), a life-threatening immune disorder. Launchis expected in Q4 2026 with logistical hurdles limiting uptake.

Aurora Therapeutics launched with the goal of scaling personalized geneediting treatments for rare diseases, building on recent proof-of-conceptsuccesses such as bespoke CRISPR therapies designed for individual patients.The company aims to industrialize this approach by using a platform modelleveraging base editing and modular guide RNAs to rapidly create multipletherapies targeting different mutations within the same disease (e.g., PKU).Aurora is poised to rely on the new FDA plausible mechanism framework.

The FDA placed clinical holds on multiple rare disease gene therapy programsin early 2026, including REGENXBIO’s RGX-111 and RGX-121, and Intellia’sCRISPR-based therapy, which has been resumed. These events highlightincreasing regulatory caution around long-term safety risks associated withgene editing and viral vector-based therapies. Recent data suggests ~84% ofinvestors have reduced or paused investment in the space, with many biotechsstruggling to raise capital as regulatory expectations become less predictable.

A coalition of patient groups and biotech executives recently penned a letterurging the Trump administration to “restore regulatory clarity” for rare diseasetherapies, citing growing concern over shifting and inconsistent FDA expectations.Despite new initiatives aimed at accelerating development, recent regulatorydecisions have introduced uncertainty, with similar therapies facing differingrequirements around trial design, surrogate endpoints, and evidence thresholds.Stakeholders note that this lack of predictability is complicating developmentplanning and increasing risk for sponsors navigating already complex rare diseaseprograms.

Ultragenyx announced that the FDA has accepted the resubmission of its BLAfor UX111, an AAV9 gene therapy for Sanfilippo syndrome type A which is arare, fatal neurodegenerative disorder with no approved treatments. Theresubmission follows a 2025 CRL driven by manufacturing and facilityconcerns, and now includes updated long-term clinical data demonstratingdurable neurologic and biomarker improvements. The application is underPriority Review, with a potential decision expected in Q3 2026.


Get expert analysis, industry trends, and exclusive updates delivered straight to your inbox. Stay ahead of the curve with valuable insights that drive success.